AID/APOBEC family represents a unique group of enzymes that function as DNA and RNA mutators. They can edit nucleotide sequence of DNA and/or RNA molecules thanks their ability to deaminate cytidine or its modified derivatives. In recent years, the detection of RNA/DNA editing events have been greatly improved, thanks the availability of high-throughput sequencing technologies. However, still bioinformatics analysis of next-generation sequencing (NGS) data sets remain a challenging task. Several pipelines are available for the detection of RNA editing events and experimentally induced mutations in genomic DNA. Accurate bioinformatics tools are essential to ensure the correct identification of edited sites. Below we present some of the most useful tools, given that in vitro activity of APOBECs causes slight nucleotide changes in RNA, and more global changes in DNA
Bioinformatics tools
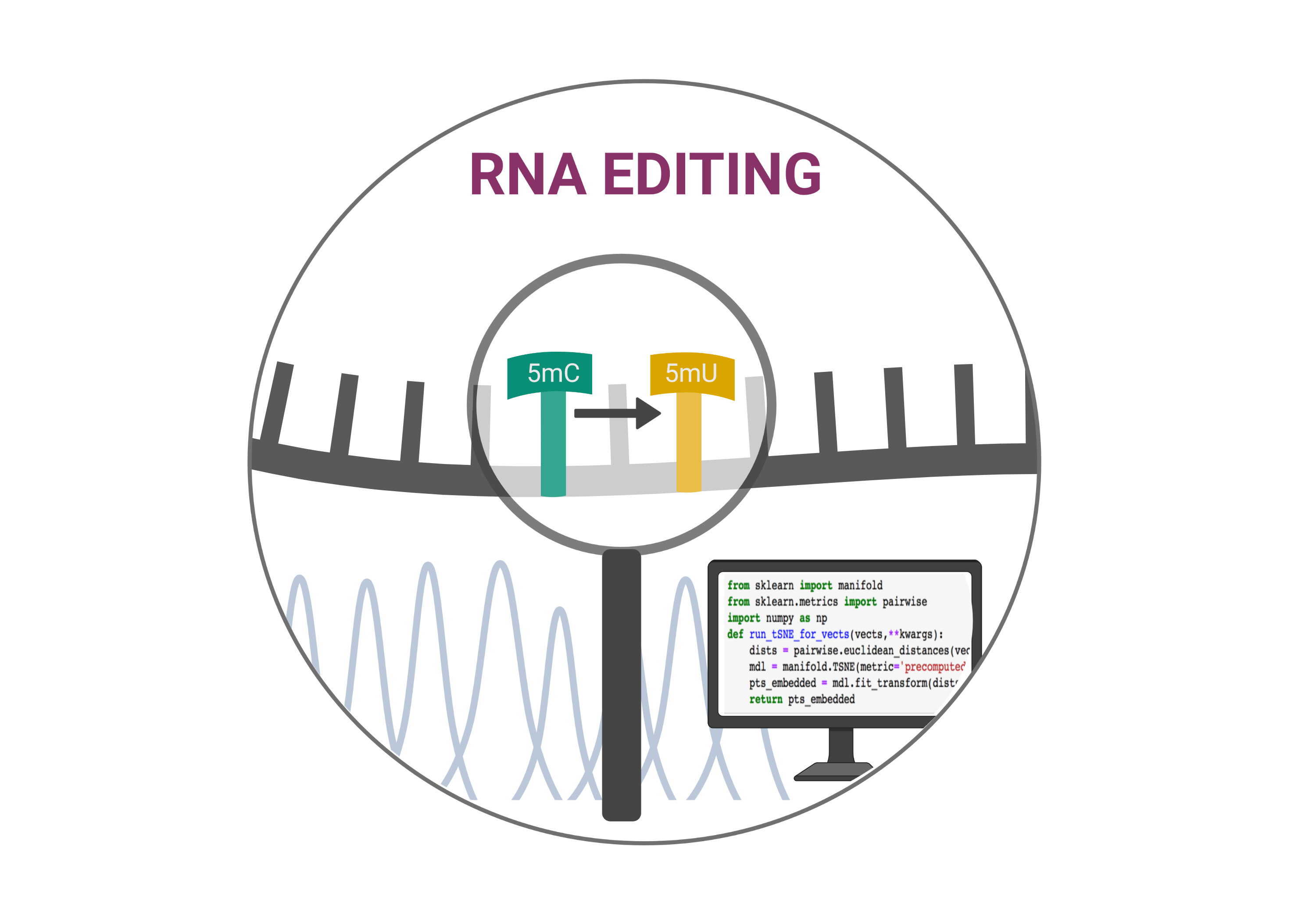
Sequencing RNA fragments produces data composed of millions of reads. Bioinforcmatics tools are able to systematically exploit these data allowing for an efficient detection of RNA editing sites in a genome-wide scale. This analysis comprises three major steps: RNA-seq data processing, RNA read alignment, and RNA editing site detection (also known as variant calling). In principle, any tools that can identify single-nucleotide variants, such as SAMtools/BCFtools and GATK, are capable of detecting RNA editing events. However, accurate identification of editing events is more challenging. SNPs and sequencing or read mapping errors far outweigh the editing signal. Therefore the bioinformatics tools must be able to distinguish the editing events from these noises. In recent years, a number of sophisticated pipelines dedicated to transcriptome-wide identification of editing events have been developed. Some of them allows for detection of RNA editing using matched RNA- and DNA-Seq data, while the others have been developed in order to identify RNA editing sites in the absence of DNA sequencing data. The first group is particularly useful in the case of C to U editing in in vitro experiments using recombinant APOBECs…
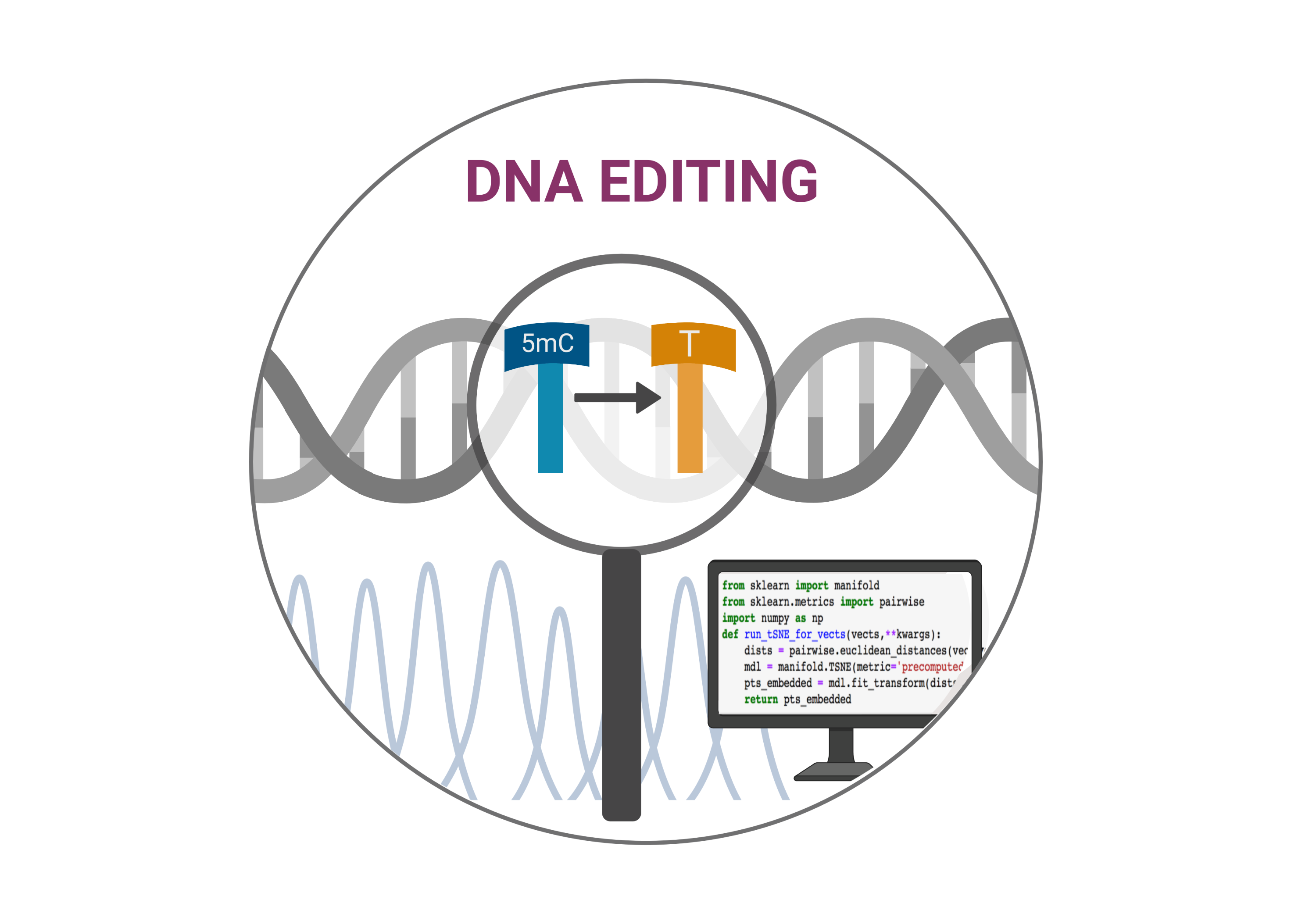
Bisulfite sequencing (BS-Seq) is a traditional approach that provides quantitative cytosine methylation levels in genome-wide scope and single-base resolution. A number of bioinformatic pipelines to exploit BS-Seq data sets have been recently developed. The goal of these pipelines is to map sequencing reads, calculate methylation levels, and distinguish differentially methylated positions and/or regions. The experimental part of BS-Seq is based on bisulfite conversion. Treatment of DNA with bisulfite converts cytosine residues to uracil in the reaction of chemical deamination, but leaves 5mC and 5hmC residues unaffected. Treatment of DNA with APOBECs results in enzymatic deamination, giving similar (but inverted) effects to BS-Seq. Consequently, in the case of APOBEC-assisted sequencing, similar bioinformatic pipelines to BS-Seq can be applied. For example Bismark software is a flexible tool for the time-efficient analysis of BS-Seq data which performs both read mapping and methylation calling in a single step. Recently, the software has been successfully applied in Tet-assisted Bisulfite Sequencing (TAB-Seq), for 5hmC mapping on a genome-wide scale (Yu et al. 2013) and in long-read enzymatic modification sequencing (LR-EM-seq) for 5mC…
Detecting RNA-Editing Events, Detecting DNA Deamination
Detecting RNA-Editing Events
In recent years, a number of software packages dedicated to transcriptome-wide identification of editing events have been developed. Most of them involve statistical models, filtering approaches, and integration of additional genomic information to improve the accuracy of editing detection and quantification. Some of these methods utilize RNA and matched DNA sequencing data from the same sample in order to reduce the identification of single-nucleotide polymorphisms and somatic mutations as editing events.
- REDItools consist of a set of scripts written in the Python language, available under the MIT license at https://github.com/BioinfoUNIBA/REDItools. REDItools are not organism oriented and work with RNA-Seq and DNA-Seq data from any sequencing platform. REDItools include three main scripts: REDItoolDnaRNA.py detects RNA editing candidates by comparing pre-aligned RNA-Seq and DNA-Seq reads; REDItoolKnown.py allows to explore the RNA editing potential of RNA-Seq experiments by looking at known events only; REDItoolDenovo.py performs the de novo detection of RNA editing candidates using RNA-Seq data alone. REDItools require inputs in BAM format, and results are provided in tab-formatted tables facilitating downstream analyses. It is highly flexible, including a variety of filters and quality checks, and may provide reliable sets of editing candidate sites.
- RES-scanner is a software package written in the Perl programming language, available under the GPL v3 license at https://github.com/ZhangLabSZ/RES-Scanner. RES-scanner provides a complete pipeline from raw sequencing reads to final editing sites and addresses read mapping, homozygous genotype calling, de novo RNA-editing site identification and annotation for any species with matching RNA-seq and DNA-seq data. In contrast to REDItools, it is equipped with statistical models (Bayesian and Binomial) to infer the reliability of homozygous genotypes derived from DNA-seq data and to distinguish RNA-editing sites from sequencing errors by assigning a p-value to each RNA-editing candidate. It runs faster than REDItools and is designed to run multiple samples in parallel.
We also recommend DARNED database (https://darned.ucc.ie) – a collection of annotated C-to-U and A-to-I editing sites in human, mouse and fly.


Detecting DNA Deamination
The processing of BS-Seq data and sequencing data after APOBEC treatment is challenging due to deamination reaction, which reduces significantly sequence complexity. Because conversion occurs only at C/5mC and not at G, strands are not complementary. These are the main concerns that computational tools must deal with, which is different compared with tools for regular DNA sequencing. A high quality of sequencing reads is crucial to get a good alignment and to correct methylation scores. Raw reads should be first trimmed using for example Cutadapt or Trim Galore software (https://github.com/FelixKrueger/TrimGalore) to remove adapter sequences and low-quality bases from the 3′end. Incorrectly converted reads should be discarded. Next, trimmed sequencing reads are aligned to the reference genome. To do this, two types of algorithms are available: wild-card or three-letter. The wild-card algorithm substitutes Cs with Ys in the reference genome, so reads can be aligned with both, Cs and Ts (tools: LAST, BSMAP, RRBSMAP, and Pash). The three-letter algorithm converts all Cs into Ts, both in the reference genome and in the reads (tools: Bismark, BRAT-BW, BS Seeker 3). Due to the asymmetrical alignments and non-complementarity, post-alignment tools (such as BSPAT and SAAP-RRBS ) are also useful. The aligned reads can be subjected to postprocessing quality control steps. E.g. PCR duplicates should be discarded, end-repair errors should be reduced, and reads that contain excessive cytosines in non-CpG context should be removed to reduce nonconversion errors. The remaining good quality alignments can be used for cytosine methylation and hydroxymethylation calling by e.g. Bismark methylation extractor.
- Bismark is a software package for the time-efficient analysis of BS-Seq data which performs both read mapping and methylation calling. Bismark is available under the GNU GPLv3+ licence at www.bioinformatics.bbsrc.ac.uk/projects/bismark/. The output of Bismark is easy to interpret and is intended to be analysed directly by the researcher performing the experiment.
This project has received funding from the National Centre for Research and Development under the LIDERIX Programme
„Mapping of cytosine modifications in nucleic acids using AID/APOBEC enzymes”; Project no: LIDER/30/0111/L-9/17/NCBR/2018